Your privacy is very important to us. When you visit our website, please agree to the use of all cookies. For more information about personal data processing, please go to Privacy Policy.






HuidaGene team of scientists collaborate to discover potential new treatment for Duchenne muscular dystrophy





Approximately 10% of monogenic diseases are caused by nonsense point mutations, which generate premature termination codons (PTCs) and lead to protein truncation and nonsense-mediated decay of mutant mRNAs, about 50% of which can be reversed by A-G conversion of adenine base editors (ABEs) to reverse the A-G transition. Specifically, A-G conversion can restore protein expression by converting PTCs to non-terminating codons, thus enabling PTCs to be read-through.
On December 13, 2022, the Journal of Clinical Investigation published an online article titled "Mini-dCas13X-mediated RNA editing restores dystrophin expression in a humanized mouse model of Duchenne muscular dystrophy" (IF=19), which is a collaboration between HuidaGene, the Center of Excellence in Brain Science and Intelligent Technology of the Chinese Academy of Sciences and Fujian Medical University. The study showed that HuidaGene's proprietary Mini-dCas13x (13e)-mediated RNA base editing (Mini-dCas13x RNA base editing, mxABE)¹ can efficiently restore the myasthenic protein (Dystrophin) expression in the humanized DMD mouse model of Duchenne muscular dystrophy. expression.
This groundbreaking research is a collaboration between HuidaGene Chief Scientific Advisor and Founder Yang Hui's research group, Professor Wang Liao and Professor Chen Wanjin's team at Fujian Medical University, Xu Chunlong's group at Lingang Laboratory/Shanghai Center for Brain Science and Brain-like Research, the Center for Excellence in Brain Science and Intelligent Technology at the Chinese Academy of Sciences, and the Institute of Agricultural Genome Research at the Chinese Academy of Agricultural Sciences. HuidaGene and the Brain Intelligence Center are the first authors of the paper; Dr. Guoling Li of the current company's Pipeline Development Department and Dr. Qingquan Xiao of the Innovation Research Institute are the co-first authors. Dr. Xing Wang, Haoxiang Wang and Linyu Shi also made important contributions to the study.
Although base editors derived from the cleavage enzyme Cas9 have been shown to correct some single gene mutations, these proteins are usually too large for effective in vivo delivery through a single AAV vector (maximum load size is only 4.7 kb).2 RNA base editing tools with smaller size, reversibility and flexibility compared to other large size base editing tools have become the focus of much development. tools have been the focus of substantial development, giving rise to REPAIR, RESTORE, LEAPER, mxABE, and other editing systems. A recent study has shown that RNA base editors can indeed restore protein expression terminated by nonsense mutations. However, this study achieved only limited functional protein expression, and no remission of impaired muscle function or restoration of motor function was observed in treated mice.3 Nevertheless, these studies suggest that RNA editing systems can target nonsense mutation-induced PTCs, implying that RNA editing may be a safer, more effective therapeutic route for treating monogenic diseases.
Among the monogenic degenerative muscle diseases, Duchenne muscular dystrophy (DMD) is the second most common inherited muscle disease. It affects an estimated 1 in 3,500-5,000 male births. There is still no effective clinical treatment for DMD, and the majority of patients have a poor prognosis and a high social burden. Glucocorticoids, as the only drug with evidence-based medical evidence recommended by current guidelines, are only able to delay the progression of the disease to a certain extent and cannot reverse the catastrophic course of DMD, although they are indicated for patients with all mutation types of DMD. Statistically, 60-70% of DMD disease is caused by exon deletion of the DMD gene. However, about 10% of cases are caused by nonsense point mutations that introduce PTCs in DMD, of which 40% are UAG, 39% are UGA, and 21% are UAA PTCs. since DMD is caused by the lack of functional dystrophin protein, restoring the function or expression of dystrophin protein at the genetic or transcriptional level is the most effective The most effective and fundamental therapeutic approach is to restore the function or expression of dystrophin protein at the genetic or transcriptional level.
Previously, a patient with progressive symmetric muscle weakness, pseudohypertrophy of gastrocnemius muscle, elevated serum creatine myokinesis and myogenic damage on electromyography was admitted to the First Hospital of Fujian Medical University, a collaborating institution. further validated its pathogenicity. Since most cells do not express the DMD gene, the authors screened for relevant DMD nonsense mutant loci using an exogenous reporter vector carrying the mutant site and found up to 84% A-to-G editing efficiency in humanized DMD mice by intramuscular spot injection of a single adeno-associated virus (AAV9)-packed mxABE system, which is in contrast to previous studies using other RNA editing modalities compared to the efficiencies reported in previous studies using other RNA editing modalities by at least 20-fold.
To explore the possibility of curing patients with DMD, the authors verified protein recovery levels and functional recovery by systemic injection of mxABE into a humanized mouse model. It was found that systemic administration of mxABE system via AAV restored dystrophin protein expression in diaphragm, tibialis anterior and cardiac muscles to an average of 37%, 6% and 54% of WT levels, respectively, and significantly restored motor function in mice. In conclusion, this study successfully achieved dystrophin protein expression in humanized DMD mice using the more efficient and smaller editing mxABE system developed by HuidaGene, and significantly restored their motor function. The completion of this project fully demonstrates the strength of Pfizer's world-leading gene mouse construction platform, which can rapidly and efficiently realize the construction of various disease models; meanwhile, the advanced gene editing technology is used to conduct safety and efficacy assessment related to new drug development in animals, which provides a powerful option for the subsequent pipeline development and treatment of related genetic diseases.
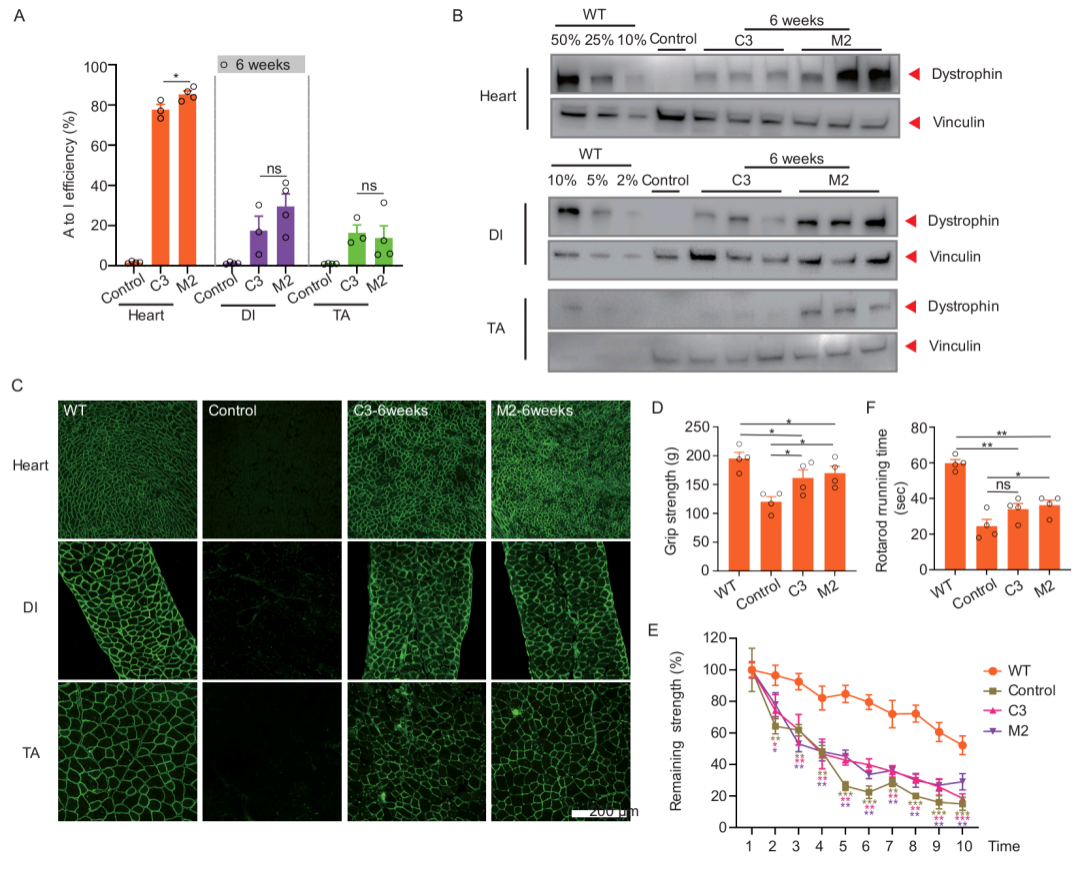
Figure 1: Recovery of humanized DMD mice detected 6 weeks after systematic injection of mxABE
1. Xu C, Zhou Y, Xiao Q, He B, Geng G, Wang Z, et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nature methods. 2021;18(5):499-506.
2. Chemello F, Chai AC, Li H, Rodriguez-Caycedo C, Sanchez-Ortiz E, Atmanli A, et al. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Science Advance. 2021;7(18):eabg4910
3. Katrekar D, Chen G, Meluzzi D, Ganesh A, Worlikar A, Shih YR, et al. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nature methods. 2019;16(3):239-42.
recommendations
-
Dec 12,2024
HuidaGene Therapeutics Initiates M.U.S.C.L.E. Clinical Trial of HG302 for Duchenne Muscular Dystrophy and Completes First Patient Dosed
-
Apr 11,2025
HuidaGene at CRISPR MEDiCiNE 2025: A Celebration of Progress, Promise, and Patients
-
Nov 04,2024
HuidaGene Therapeutics Receives the First-Ever FDA Clearance of CRISPR/Cas13 RNA-Editing HG202 for Macular Degeneration
-
Dec 06,2024
HuidaGene Therapeutics Announced First Patient Dosed in the HERO Clinical Trial of HG204 for MECP2 Duplication Syndrome