Your privacy is very important to us. When you visit our website, please agree to the use of all cookies. For more information about personal data processing, please go to Privacy Policy.






What is Rhodopsin-mediated Autosomal Dominant Retinitis Pigmentosa?
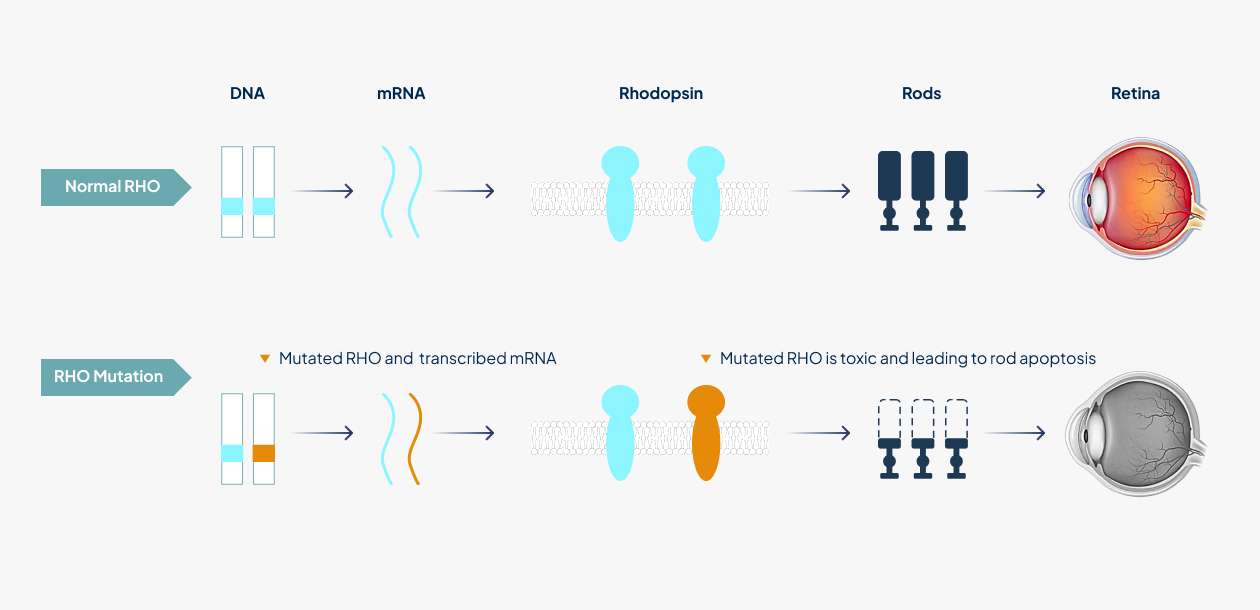
Retinitis pigmentosa (RP), affecting 1 in 3,000 - 7,000 people worldwide, is a genetically and phenotypically heterogeneous group of diseases which is characterized by the death of rod photoreceptor cells leading to defects in dark adaptation and night blindness. Rhodopsin-mediated autosomal dominant retinitis pigmentosa (RHO-adRP) is a hereditary degenerative disorder in which mutations in the gene encoding RHO, the light-sensitive G protein-coupled receptor involved in phototransduction in rods, lead to progressive loss of rods and subsequently cones in the retina, afterwards leading to blindness in later life¹. More than 150 gain-of-function mutations within the rhodopsin (RHO) gene account for approximately 25% of autosomal dominantly inherited RP cases.
Worldwide, there are an estimated 175,000 people2 living with RHO-adRP.
1. Da Meng. et al. Molecular Therapy. 2020. Therapy in Rhodopsin-Mediated Autosomal Dominant Retinitis Pigmentosa
2. Lewin AS. et al. Cold Spring Harb Perspect Med. 2022. Gene Therapy for Rhodopsin Mutations.
-
estimated 175,000living with RHO-adRP

What Are the Symptoms Associated with RHO-adRP?
Clinical phenotypes are diverse, ranging from mild night blindness (nyctalopia) to severe visual impairments. In the initial stages of the disease process, rod photoreceptors start to die causing night blindness. Patients began to experience difficulty seeing in dim light and leading to difficulties with driving at night or entering darkened rooms. Cone photoreceptors are progressively lost and continuous loss of photoreceptors leads to loss of peripheral vision termed “tunnel vision”, whereby only the central vision is preserved. Central vision is finally lost in the late stages of the disease, resulting in total blindness¹.
More than 150 mutations in RHO gene cause adRP and can fall into two classes by clinical phenotype² .
Class A patients (possessing mutations R135G, R135L, R135W, V345L, and P347L) lose rod function over the entirety of the retina and experience onset of night blindness earlier in life.
Class B patients (possessing mutations T17M, P23H, T58R, V87D, G106R, D190G, G51A, Q64ter, and Q344ter) demonstrate a milder phenotype, including normal rod activation kinetics and preserved rod outer segment length with abnormalities in the rod visual cycle that are mutation specific.

1. Da Meng. et al. Molecular Therapy. 2020. Therapy in Rhodopsin-Mediated Autosomal Dominant Retinitis Pigmentosa. 2. A V Cideciyan, et al. Proc Natl Acad Sci U S A.1998. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man.
How is RHO-adRP Diagnosed?
Clinical assessment of patients with RP starts with complete past medical history and family history, such as poor vision and night blindness, and then confirmed by combination of clinical examinations including psychophysical tests, slit lamp examination, fundus examination, electroretinogram (ERG), fundus autofluorescence, and spectral domain optical coherence tomography (OCT)¹ . Furthermore, genetic testing can be used to distinguish whether RP is medicated by RHO mutations.

1. Jose´ -Alain Sahel. et al. Cold Spring Harb Perspect Med. 2015. Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations.

What Are the Treatment Options for RHO-adRP?
-
1.RHO-adRP diagnosed
There is currently no effective treatment or cure for RHO-adRP, although multiple approaches are being studied, like stem cell or retinal tissue transplantation, nutritional supplementations (such as vitamin A, docosahexaenoic acid (DHA), and lutein but with limited and controversial results), retinal implants, as well as targeted and non-targeted gene therapies1. CRISPR-based gene editing technologies (such as HuidaGene and other companies) are now increasingly being developed as therapeutics for the disease to permanently correct the underlying mutations.
Previous : HG202(nAMD)
Next : No more